Adolescent-Onset and Adult-Onset Vitamin-Responsive Neurogenetic Diseases
Daniele Mandia, JAMA Neurol. Published online January 11, 2021.
Importance Vitamin-responsive inherited diseases are among the rare genetic disorders with a specific pharmacological treatment. Many of these conditions have a prominent neurological phenotype that is mainly reported in children. Being rare and often strikingly different in adult-onset forms, they are still poorly known in the medical fields specific to adults.
Observation This article reviews all articles reporting cases of patients with a genetically confirmed inherited vitamin-responsive neurological disease and neurological onset after the age of 10 years. On this basis, 24 different diseases are described, involving vitamins A, B1, B2, B3, B6, B8, B9, B12, E, and tetrahydrobiopterin (BH4). Information such as clinical symptoms, disease course, imaging studies, biochemical alterations, and response to treatment present an overall picture of these patients.
Conclusions and Relevance Vitamin-responsive neurogenetic diseases represent a group of rare conditions that are probably underdiagnosed in adults and may have a dramatic response to treatment when started early in the course of the disease. In this review, main features of the adult-onset forms are defined and simple key messages are provided to help identify clinical situations when specific diagnostic tests should be performed and/or vitamins should be promptly administered.
Introduction
Adolescent-onset and adult-onset neurogenetic diseases are a heterogeneous group of disabling conditions, with extremely diverse phenotypes, numerous genes involved, and for the most part, no specific treatments. However, some of these diseases can respond to specific vitamin supplementation.1 Vitamins are essential micronutrients produced in insufficient quantities or not produced by humans, who nonetheless have all the cellular machinery to transform them into active forms: the enzyme cofactors.
[b]Vitamin supplementation is easily available, usually without adverse effects, and dramatically efficient, especially if given early in the course of these diseases.[/b] Therefore, rapid identification of vitamin-responsive diseases is crucial for neurologists. This review aims at clarifying the adolescent-onset and adult-onset phenotypes of vitamin-responsive neurogenetic diseases, which are often different from early-onset forms. We also suggest clinical situations in which specific diagnostic tests should be performed and/or vitamins should be administered as a therapeutic trial.
Methods
Through a review process, we identified 24 neurogenetic diseases, characterized by possible neurological symptoms that started after the age of 10 years and a clear response to vitamin supplementation for at least a subset of patients. These diseases, involving vitamins A, B1, B2, B3, B6, B8, B9, B12, E, and tetrahydrobiopterin (BH4) coenzyme, are listed in the eAppendix and eTable 1 in the Supplement, among all other genetic vitamin-responsive diseases. We chose this age threshold (referred in the text as adult-onset) since, in our experience, inherited metabolic diseases starting after the age of 10 years show phenotypes usually similar to those of patients with later onset and are often different from earlier-onset forms.2-4 Despite that BH4 is not properly a vitamin, it is an organic compound that acts similarly to vitamin-derived cofactors,5 which explains why we considered BH4-associated conditions to be part of vitamin-responsive diseases.6
Review
Following our analysis of the literature, we divided the pathophysiology of vitamin-responsive diseases in 4 major groups: those with
(1) impaired cofactor synthesis from the vitamins;
(2) impaired transport (intestinal absorption, blood transport, or cellular uptake);
(3) a defect of an enzyme that requires a vitamin-derived cofactor; and
(4) a secondary vitamin defect (attributable to abnormalities that are not associated with a gene defect directly involved in vitamin metabolism).
Depending on pathophysiology, optimal supplementation efficiency may need a vitamin (or its derived cofactor) at various pharmacological dosages and routes of administration. As for disease type 3, efficiency depends on the presence of vitamin-responsive sequence variations, often found in the adult-onset form of the disease,7-10 whereas for disease type 4, efficiency can be very modest, in that a vitamin defect may only represent an epiphenomenon of the disease.
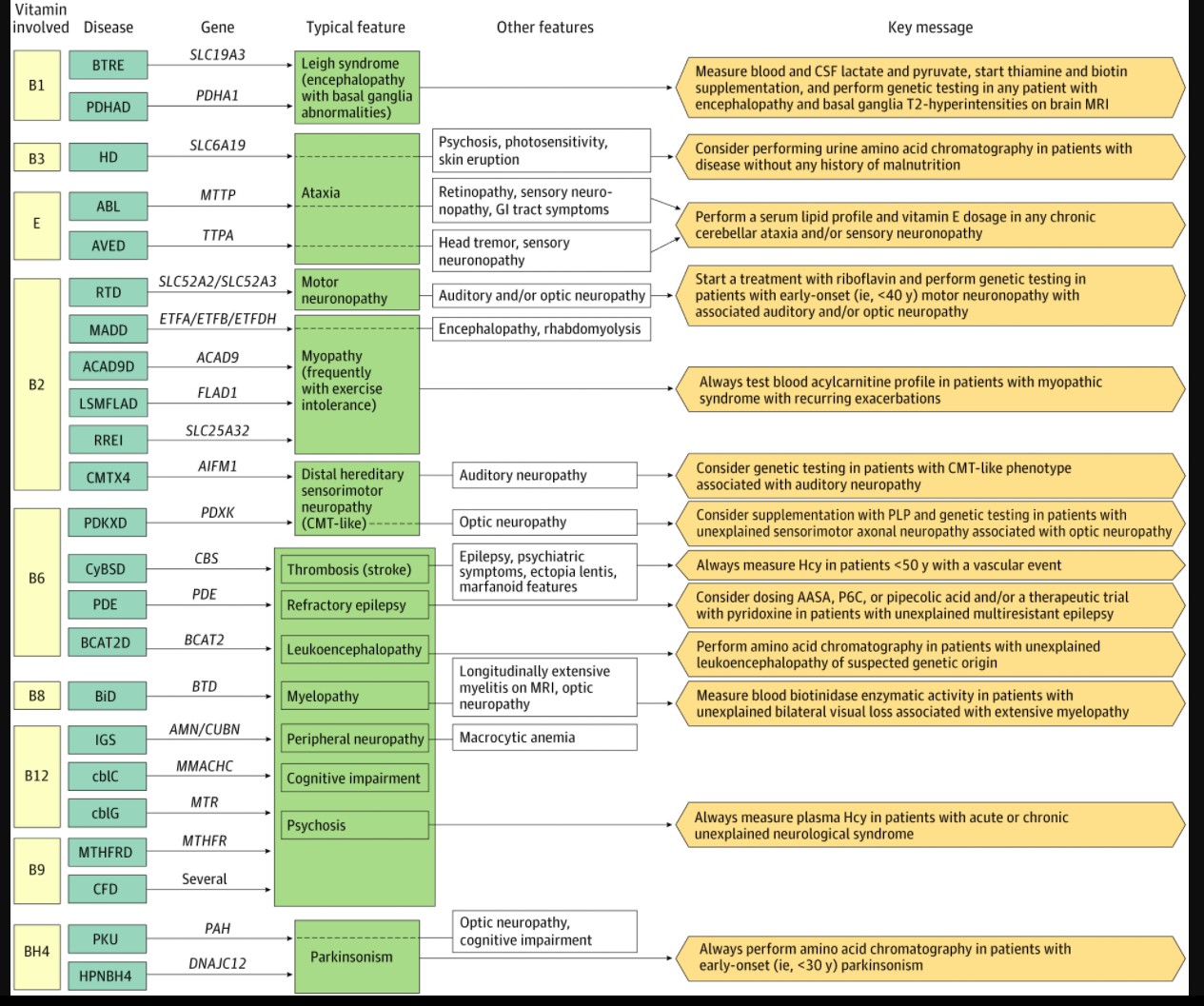
Considering that overlapping phenotypes can result from different genetic diseases, we have chosen to highlight this clinicobiological correlation as a pragmatic approach to these numerous and complex conditions. The collected relevant data on adult-onset neurogenetic diseases are summarized in the eAppendix and eTable 2 in the Supplement (clinical manifestations, diagnostic tests, and therapeutic options, with their expected efficiency), eTable 3 in the Supplement, and Figure 2, which also suggests associated diagnostic approaches.
Thiamin Defects and Targeting of the Basal Ganglia
Wernicke encephalopathy is an acquired clinical condition caused by a thiamin (or vitamin B1) deficiency, often in a context of malnutrition and/or disordered alcohol use. The encephalopathy is usually associated with peculiar features, such as cerebellar syndrome, seizures, ophthalmoplegia, and/or ptosis.11 A close phenotype can be seen in adults in 2 thiamin-associated neurogenetic diseases: biotin-thiamin–responsive encephalopathy (BTRE) and E1-α pyruvate dehydrogenase deficiency (PDHAD). These 2 conditions are treatable causes of the genetically heterogeneous Leigh syndrome.12 In BTRE, thiamin transport into cytosol is reduced as a result of a defect of the thiamin transporter type 2 (coded by solute carrier family 19 member 3 gene [SLC19A3]), leading to a low intracellular thiamin level, whereas the blood thiamin level is normal.9 In PDHAD, the abnormal pyruvate dehydrogenase E1-α subunit (coded by pyruvate dehydrogenase E1-α subunit gene [PDHA1]) prevents the correct binding of its cofactor thiamin pyrophosphate, the active form of thiamin13 (Figure 3). This malfunction has repercussions on the tricarboxylic acid cycle, with reduced production of acetyl-CoA, and leads to a switch toward anaerobic production of energy and eventually to lactic acidosis, which is also present in BTRE. In addition to high lactate levels, high pyruvate levels in blood and the cerebrospinal fluid are characteristic features of PDHAD. As other brain-energetic diseases, circumstances of increased metabolic demand, such as febrile episodes, surgery, or traumas, can trigger the encephalopathic episodes. However, progressively worsening chronic symptoms, such as dystonia (present in the acute phase as well), can also occur. Brain magnetic resonance imaging typically shows T2 hyperintensities of caudate heads and putamens (Figure 4A and B for BTRE), as in Leigh syndrome, but also mesial thalami and periacqueductal gray matter, as in Wernicke encephalopathy.14 In BTRE, a cortical involvement with scattered foci of T2 hyperintensities has also been described in the acute phases of the disease.15,16 Despite thiamin being the mainstay of the treatment in both diseases, in BTRE, a supplement with biotin is also recommended, which probably increases SLCA19A3 expression through histone biotinylation.17 A ketogenic diet is also prescribed in PDHAD, since it increases the liver production of ketone bodies, an alternative energy substrate to glucose for the generation of acetyl-CoA in the central nervous system.18
Riboflavin in Muscle and Nerve Diseases
Riboflavin (or vitamin B2) is a water-soluble and photosensitive compound at the origin of flavin mononucleotide and flavin adenine dinucleotide (FAD). These 2 cofactors act as electron transporters in numerous biochemical reactions, including the mitochondrial respiratory chain, involving proteins called electron transfer flavoproteins (ETF). Acquired riboflavin deficiency is rare, and it can be attributable to inadequate diet (especially when milk consumption is low), disordered alcohol use (in that ethanol blocks riboflavin receptors at the intestinal level), and infantile phototherapy (used to treat hyperbilirubinemia).19 The genetic diseases involving riboflavin described here are defects of (1) the cellular uptake in riboflavin-transporter deficiency (RTD), (2) mitochondrial fatty acids β-oxidation, seen in multiple acyl-coA deficiency (MADD; defects of ETF and ETF ubiquinone-oxidoreductase), and quite similar diseases more recently described (acyl-coA dehydrogenase 9 deficiency, lipid storage myopathy attributable to FAD synthetase deficiency, and riboflavin-responsive exercise intolerance attributable to a defect of the mitochondrial FAD transporter); and (3) a FAD-dependent oxidoreductase in Charcot-Marie-Tooth X-linked disease type 4 (CMTX4) (Figure 3). In all these conditions, the prominent feature of adult-onset forms is a peripheral nerve or muscle disorder. As such, patients with RTD have a purely motor (or, rarely, sensorimotor) neuronopathy with early bulbar features that may mimic amyotrophic lateral sclerosis; patients with CMTX4 have Charcot-Marie-Tooth–like disease (distal sensorimotor hereditary peripheral neuropathy) with chronic gait difficulties, distal amyotrophy and weakness, areflexia, and pes cavus. Patients with late-onset MADD usually present with myopathic signs, proximal weakness, and exercise intolerance. Interestingly, in both RTD and CMTX4, auditory neuropathy is present early in the course of the disease. Optic neuropathy can also be associated in patients with RTD. Multiple acyl-coA deficiency can also present with acute decompensations with lactic acidosis and rhabdomyolysis in cases of increased energetic demand (eg, intense physical exercise, fasting, infections), heavy alcohol consumption, vomiting, or diarrhea. Metabolic investigations are very useful in MADD and associated diseases, in which the blood acylcarnitine profile is always abnormal, with accumulation of all-chain length acylcarnitines, whereas this feature is not constant in RTD. Riboflavin supplementation usually lead to a rapid and dramatic improvement in MADD and associated diseases, whereas it usually takes several months to show clinical effects in RTD and CMTX4.
The Various and Complex Clinical Pictures of Pyridoxine, Folate, and Cbl Defects
These 3 vitamins, also named B6, B9, and B12, respectively, are routinely measured in blood and supplemented because they are often found to be responsible for acquired deficiencies causing neurological symptoms.20 Pyridoxine acts as the cofactor of the enzyme cystathionine β-synthase (CyBS) that ensures the transsulfuration of the homocysteine (Hcy) to cystathionine for its degradation.
Folate and Cbl are also directly involved in Hcy metabolism, both being necessary for its remethylation to Met (Figure 1). Therefore, a genetic defect in the biochemical pathway of each of these vitamins may lead to severe hyperhomocysteinemia (usually greater than 100 μmol/L), associated with high blood Met levels in individuals with CyBS deficiency or low Met levels in those with folate-associated and Cbl-associated diseases. On the other hand, blood vitamin levels are often normal. Pyridoxine is also involved in numerous other enzymatic reactions from various metabolic pathways (including production of neurotransmitters, such as γ-aminobutyric acid), explaining the very diverse manifestations of pyridoxine-associated neurogenetic diseases in adults: young-onset (ie, <50 years) cerebrovascular thrombosis in CyBS deficiency associated with ectopia lentis and marfanoid features; multidrug-resistant epilepsy in pyridoxine-dependent epilepsies; severe leukoencephalopathy with unspecific mild symptoms in branched-chain amino acid transferase 2 deficiency; and Charcot-Marie-Tooth–like axonal peripheral neuropathy with bilateral optic neuropathy in pyridoxal kinase deficiency. Importantly, despite that pyridoxine remains the mainstay of treatment of these conditions, dosages greater than 200 mg/day may cause serious adverse effects, the most important being sensory neuronopathy.21
An acquired folate deficiency, usually attributable to inappropriate diet, drugs (eg, methotrexate), or overconsumption during pregnancy, can result in a complex clinical picture, possibly involving both central and peripheral nervous systems, with acute and/or chronic cognitive, psychiatric, and/or motor symptoms associated with a leukoencephalopathy (Figure 4D), a myelopathy (occasionally with T2 hyperintensities on magnetic resonance imaging), and/or an axonal polyneuropathy.22 The most frequent genetic defect of folate metabolism, 5,10-methylenetetrahydrofolate reductase deficiency, can present this broad and heterogeneous phenotype.
Another folate-associated syndrome, named cerebral folate deficiency, has been recently described in adults and presents with low folate levels in cerebrospinal fluid, as a result of impairment of transport of the active form of folate from the blood to cerebrospinal fluid at the brain-blood barrier. Cerebral folate deficiency was reported mainly in mitochondrial disorders, but a consistent part of late-onset cerebral folate deficiency, despite being suspected as being of genetic origin, remains currently unexplained.23
In addition to the metabolism of Hcy, Cbl is also involved in the production of succinyl-CoA, an intermediate of the tricarboxylic acid cycle derived from methylmalonyl-CoA (Figure 1). Methylmalonic aciduria and homocystinuria, type C is the most common genetic defect of Cbl metabolism described in adults, characterized by increased methylmalonic acid and Hcy levels. Late-onset cblC has a clinical-radiological phenotype similar to 5,10-methylenetetrahydrofolate reductase deficiency (Figure 4E), except with possible associated hemolytic-uremic syndrome and/or pulmonary arterial hypertension. Since cblC is caused by a defect of the decyanation of Cbl, cyanocobalamin (the classical pharmaceutical form of Cbl) cannot be used, and patients should be treated by hydroxycobalamin. Two other, less frequent defects of Cbl metabolism diseases have been described in adults: cblG, caused by a deficiency of methionine synthase, which is responsible for the remethylation of Hcy to Met, and Imerslund-Gräsbeck syndrome, where an impairment of gastrointestinal tract–blood transport results in Cbl deficiency.
Hartnup Disorder, the Genetic Alias of Pellagra
Niacin, or vitamin B3, is mainly known in neurology for its role in the disease pellagra, which is caused by malnutrition and has now almost disappeared, at least in higher-income countries.24 Hartnup disorder, a genetic condition caused by sequence variations in the neutral amino acid transporter B0AT1, mainly expressed in intestine and renal proximal tubules, has almost an identical clinical presentation as pellagra, associating cerebellar ataxia, psychiatric symptoms, and photosensitivity. In fact, the impairment of this transporter results in the loss of several neutral amino acids, including tryptophan, which is not only the precursor of niacin but is also involved in the production of melatonin and serotonin.
Biotinidase Deficiency: a Neuromyelitis Optica Mimic
Biotinidase is an enzyme responsible for the recycling of biotin (vitamin B8), the cofactor for human carboxylases. Biotinidase deficiency results therefore in biotin deficiency, reduction of carboxylases activity, and consequent impairment of several metabolic pathways, such as gluconeogenesis, fatty acids biosynthesis, and amino acids catabolism. As in other energetic defects (eg, BTRE, PDHAD, MADD), symptoms are often exacerbated by a stressful event, such as fever or trauma. From a clinical point of view, late forms present with bilateral optic neuropathy and longitudinally extensive myelopathy25 (Figure 4C) and therefore should be investigated in cases of seronegative or refractory neuromyelitis optica.
Vitamin E and Equilibrium
All the vitamins described until this point are water soluble, but at least 4 other vitamins are liposoluble, such as vitamin A (or retinol), the 3 forms of vitamin K (phylloquinone, menaquinone, and menadione), vitamin D (or cholecalciferol), and vitamin E (or tocopherol). Among these, only vitamins A and E, when deficient, cause neurological symptoms in adults. A dietary deficiency of these 2 vitamins is currently rare, but an impairment of lipid metabolism and/or absorption could result in a deficiency.
Abetalipoproteinemia (ABL), for example, is a genetic defect of the microsomal triglyceride transfer protein, whose main function is to transfer triglycerides to apolipoprotein B to generate very low-density lipoproteins in hepatocytes and chylomicrons in enterocytes.26 Therefore, the synthesis of chylomicrons and very low-density lipoproteins, carrying fat-soluble vitamins in blood, is impaired, leading to a reduced transport of these vitamins to the peripheral tissue. As a result, both vitamins A and E are in deficient amounts. Another disease, which only involves vitamin E, is called ataxia with vitamin E deficiency and is caused by a defect of the α-tocopherol transfer protein (α-TTP), which is responsible for its incorporation in very low-density lipoproteins, resulting in its degradation.27 In both ABL and ataxia with vitamin E deficiency, blood vitamin E levels are very low, and patients suffer from motor difficulties because of cerebellar and sensory ataxia (from involvement of posterior columns and/or sensory neuronopathy). Additionally, in patients with ABL, malabsorption syndrome (eg, chronic diarrhea, short stature, anemia) starting in childhood28,29 and nyctalopia (which is a known consequence of vitamin A deficiency on retina function)30 are often the first clinical symptoms to occur. When treatment is started at this stage, it usually helps to prevent neurological damage.31 In ataxia with vitamin E deficiency, vitamin E supplementation may improve symptoms if given early in the course of the disease.27 Importantly, although vitamin A should be administered in ABL, doses greater than 4000 UI/kg in adults can have adverse effects, such as skin changes, anemia, and hypercalcemia, whereas more than 10 000 UI/day in pregnant women can be teratogenic.32
BH4 and Parkinsonism
Parkinsonism is the clinical expression of low brain levels of dopamine neurotransmitter, generated from phenylalanine by a 3-step synthetic pathway involving phenylalanine hydroxylase and tyrosine hydroxylase enzymes, both requiring BH4 as a cofactor and a chaperone molecule, the heat-shock protein DNAJC12.33 Phenylketonuria, caused by a malfunction of phenylalanine hydroxylase, is a common genetic cause of intellectual deficiency that can be prevented by a low-protein diet, often introduced after newborn screening. Phenylketonuria occasionally shows clinical and biochemical response to the administration of BH4. Rarely, when starting at adolescent or adult ages, clinical manifestations include parkinsonism often associated with additional features, such as visual acuity loss, cognitive impairment, and/or leukoencephalopathy. Responders and nonresponders have the same phenotype but different genotypes.34 Another recently discovered condition, caused by sequence variations in DnaJ heat shock protein family member C12 gene (DNAJC12), causes a nonprogressive parkinsonism and mild cognitive impairment in adults, with a dramatic response to BH4.35
Limitations
Inorganic enzyme cofactors (mainly trace minerals) are outside the scope of this review. Associated diseases were reported elsewhere.36-40
Conclusions
Neurogenetic vitamin-responsive diseases are rare but probably underdiagnosed. A better clinical knowledge of this subject could help the neurologist in anticipating diagnosis and treatment administration and therefore dramatically improve patients’ quality of life. Clinically oriented gene panels, whole-exome sequencing, and whole-genome sequencing are now valuable tools to facilitate the diagnostic process. However, in several clinical situations described in this review, a first-line or parallel biochemical diagnostic approach is still valid for several reasons. First, some clinical situations do not initially suggest a genetic cause, prompting exhaustive investigations to find an acquired causative mechanism that should include some metabolic investigations. Second, the results of genetic testing may delay vitamin administration in situations where early treatment is important. Third, the genetic testing may miss some variants difficult to identify or interpret, especially with whole-exome and whole-genome sequencing. Finally, the genetic tests are not easily available worldwide, whereas vitamins are. As this field is rapidly expanding, it is probable that some vitamin-responsive diseases supposed to be restricted to children will be reported in adults in the future, sometimes with an unexpected phenotype. New neurometabolic diseases are also frequently reported and may be vitamin-responsive. For this reason, we suggest to consider a vitamin supplementation trial in patients with phenotypes similar or close to a known vitamin-responsive genetic disease, even if classical biochemical and genetic investigations (including whole-exome and whole-genome sequencing) have negative results.